")
")










- Vous êtes ici :
-
Accueil

- Vision
Vision
La rétine est le tissu qui tapisse le fond de l'œil et qui reçoit, module et transmet le signal lumineux au cerveau afin d’assurer notre vision. La perturbation d'une de ces étapes rétiniennes entraîne des déficiences visuelles connues sous le nom de dystrophies rétiniennes héréditaires (DRH). Ces dystrophies de la rétine, caractérisées par la perte constante des photorécepteurs (les cellules neuronales qui captent la lumière), peuvent se manifester à tous les âges de la vie. Ces maladies invalidantes ont pour conséquence une atteinte progressive de la vision soit centrale de lecture, d’écriture, de reconnaissance des visages par atteinte des photorécepteurs de type cônes, soit périphérique avec une perte du champ visuel et des difficultés de vision de nuit par atteinte des photorécepteurs de type bâtonnets. Il est donc nécessaire d’élaborer et de valider de nouveaux traitements afin d’empêcher la progression de la maladie ou la perte des photorécepteurs, et éventuellement de restaurer, au moins en partie, la vision altérée. L’équipe Vision étudie les DRH dans leur globalité : 1) le diagnostic clinique et génétique ; 2) l’identification de nouveaux gènes et la validation de nouveaux variants génétiques (les altérations dans la séquence de l’ADN) ; 3) l’élucidation des mécanismes physiopathologiques, et 4) le développement de thérapies innovantes.
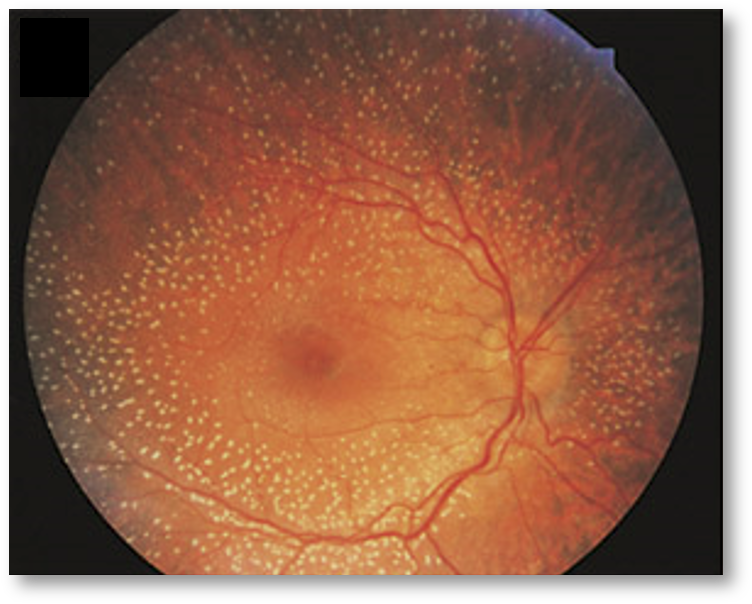
Les DRH sont des atteintes neurodégénératives de la rétine avec une prévalence de 1/3000 dans les pays industrialisés. Ce sont des maladies génétiques évolutives qui entraînent la dysfonction puis la mort cellulaire, notamment des photorécepteurs (les cellules réceptrices de la lumière) ou/et de l’épithélium pigmentaire rétinien (tissu de soutien). Ces maladies peuvent donner une atteinte rétinienne isolée ou associée à d’autres organes, et tous les modes de transmission héréditaire sont possibles.
Plus de 280 gènes sont impliqués dans des DRH et peuvent entraîner des atteintes rétiniennes isolées ou syndromiques i.e. associées à d’autres symptômes que visuels. De la même manière, plus de 200 gènes sont impliqués dans des formes isolées ou syndromiques de surdité. Depuis l'an 2000, notre groupe réalise le diagnostic moléculaire des cécités et surdités neurosensorielles. La forme syndromique la plus fréquente impliquant un phénotype oculo-auditif est le syndrome de Usher (environ 3 naissances sur 100 000) dans lequel on retrouve une surdité congénitale associée à une rétinite pigmentaire qui apparaît entre la petite enfance et l’âge adulte en fonction des gènes impliqués et des altérations moléculaires (types de variants pathogènes).
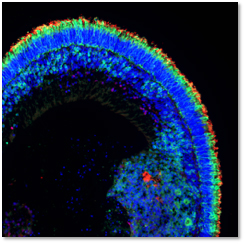
Face à la complexité et à l’hétérogénéité des DRH, il reste essentiel d’appréhender la physiopathologie (le mécanisme) de ces dystrophies et de développer des thérapies pour les traiter. Notre principal modèle de travail, mais non exclusif, est le modèle de rétine humaine issu de cellules souches pluripotentes induites (iPSCs) que nous générons à partir d’une biopsie de peau des patients prélevés dans notre centre de référence Maolya. Nous différencions les iPSCs soit en épithélium pigmentaire, soit en organoïdes rétiniens, structure en 3D contenant des photorécepteurs.
