")
")










- You are here:
-
Home

- Vision
Vision
The retina is the tissue that lines the back of the eye and receives, modulates, and transmits the light signal to the brain to ensure our vision. Disturbance of any of these retinal processes leads to visual impairments known as Inherited Retinal Dystrophies (IRDs). These retinal diseases, characterized by the progressive loss of photoreceptors (the light-sensing cells), can appear at any age. IRDs result in progressive impairment of either central vision, comprising reading, writing, and face recognition due to defective cone photoreceptors, or peripheral vision, with loss of visual field and night vision difficulties due to defective rod photoreceptors. It is therefore necessary to develop and validate new treatments to prevent disease progression or photoreceptor loss, and possibly restore, at least partly, impaired vision. The Vision team studies all aspects of IRDs: 1) clinical and genetic diagnosis; 2) identification of new genes and validation of new genetic variants (alterations in the DNA sequence); 3) elucidation of pathophysiological mechanisms, and 4) development of innovative therapies.
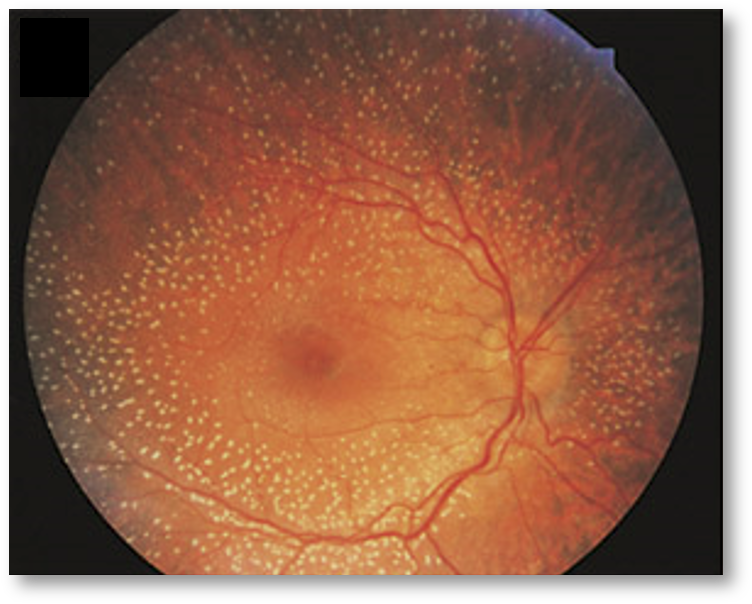
IRDs are neurodegenerative disorders of the retina with a prevalence of 1/3000 in industrialized countries. They are progressive genetic diseases that lead to dysfunction and subsequent cell death, particularly of photoreceptors (light-sensing cells) and/or the retinal pigment epithelium (supporting tissue). These diseases can result in isolated or syndromic retinal disease, and all modes of hereditary transmission are possible.
Over 280 genes are involved in IRDs and can lead to isolated or syndromic disorders, i.e., associated with symptoms other than visual impairment. Similarly, over 200 genes are implicated in isolated or syndromic forms of deafness. Since the year 2000, our group has been performing molecular diagnosis of neurosensory blindness and deafness. The most common syndromic form involving an ocular-auditory phenotype is Usher syndrome (approximately 3 births per 100,000), in which congenital deafness is associated with retinitis pigmentosa (RP) that appears between early childhood and adulthood depending on the genes involved and the molecular alterations (types of pathogenic variants).
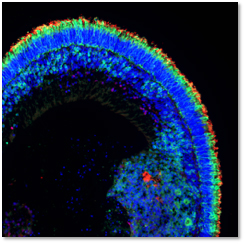
Given the complexity and heterogeneity of IRDs, it remains essential to understand the pathophysiological mechanisms underlying these dystrophies and to develop therapies to treat them. Our main, but not exclusive, working model is the human retinal model derived from induced pluripotent stem cells (iPSCs) that we generate from skin biopsies of patients within the framework of our national reference centre, Maolya. We differentiate iPSCs into either retinal pigment epithelium or 3D retinal organoids containing photoreceptors.
